
【雷特氏症候群 (RETT SYNDROME)】為一罕見的複雜性神經系統疾病,好發於女性,患孩通常在一歲以後有快速退化及發展遲緩的現象。
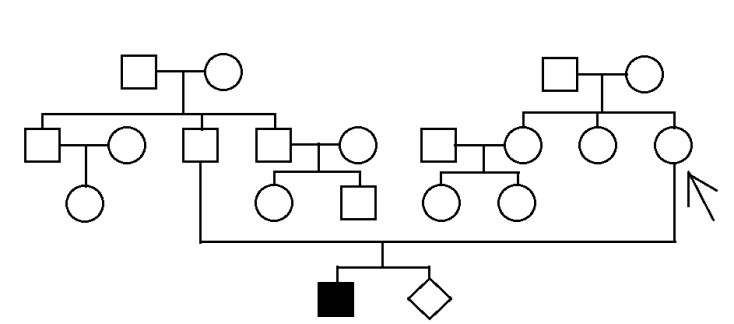
陳小姐(以下簡稱個案),2014年1月自然產娩出男嬰,產前因第一孕期風險評估為低風險,並未做非侵入性染色體檢查或羊膜穿刺染色體及晶片檢查。但孩子兩歲後發現有發展遲緩現象,至醫院檢查,基因診斷為:雷特氏症候群,具有 MECP2 基因 c.763C>T, p.Arg255*鑲嵌型之突變點位(mosaic rate: 50%)。
個案當時已懷第二胎,十分擔心會有相同症狀,故進一步基因諮詢,與醫師討論後夫妻倆決定先確診彼此是否具有同樣基因,再做後續檢查,報告結果顯示個案與先生皆未發現 MECP2 基因,後續依正常規則產檢即可,則第一胎罹患雷特氏症候群非來自遺傳而是寶寶本身基因突變所導致。
遺傳模式
雷特氏症(Rett syndrome)是神經性系統的罕見疾病,因X染色體上MECP2序列的基因突變所造成的,臨床上常被誤診為自閉症、腦性麻痺或未特定的發展性疾患。雷特氏症好發於女性,在女性新生兒的發生率約1/10,000~1/12,000,患者在一歲以前狀似正常,多數在一歲以後發病。只有極少數男孩可以存活,並且表現典型雷特氏症的症狀。
雷特氏症的特徵及發展階段:
| 第一期:早期 | 6個月∼1歲半 | 嬰兒很安靜、乖巧,目光不注視外界人物,頭圍成長趨緩。 |
| 第二期:發展明顯遲緩及退化期 | 1∼4歲 | 快速退化及發展遲緩現象,包括語言表達逐漸喪失,呈現洗手、捻手、搓手等刻板動作,偶而的將手無理由的放在背後觸摸,握緊手、張手等,有的患孩會呈現吐舌、陣發喘氣、睡眠不安或走路開始不穩,頭圍明顯成長趨緩。 |
| 第三期:幼稚園至國小年齡之穩定期 | 2∼10歲 | 呈現失用症,不會使用正常力量的肢體;躁動、愛哭。 |
| 第四期:運動退化之晚期 | 5∼25歲 | 失能,逐漸呈不能走路,有些因下肢呈強直,需坐輪椅,但認知、語言溝通及手部動作較穩定,注視他人能力仍能維持。 |
「雷特氏症」目前無法根治,只能透過藥物治療併發症,可透過音樂治療「雷特氏症」,搭配拍手等肢體動作,讓孩子學習與父母親互動,增進孩子的溝通能力;並研發各式輔具,避免因雙手搓揉,造成身體受傷。
遺傳諮詢服務,可協助病家瞭解醫療事實,包括診斷、病程及治療,瞭解遺傳形式及再發機率,以及協助病家選擇最適合個人及家庭的措施等,避免罕病再製,也能早期發現早期治療。
此為禾馨醫療個案 非經同意請勿轉載
個案圖譜







發表留言