
【甘迺迪氏症(Kennedy Disease)】是一種性聯隱性遺傳疾病,由美國的甘迺迪醫師首次於1968年完整報告而得名,因為患者的脊髓和延髓會退化、神經細胞失去功能甚至壞死,而導致肌肉萎縮無力,所以又被稱為「脊髓延髓性肌肉萎縮症」(Spinal and Bulbar Muscular Atrophy;SBMA)。
小芳(化名) 2018年4月至已育有一女,因有計劃再懷第二胎,至禾馨懷寧進行遺傳諮詢,經了解小芳爸爸下肢無力多年,原以為是糖尿病造成末梢神經病變所導致,至台大神經科就診醫師懷疑甘迺迪氏症(Kennedy Disease),進一步基因檢測結果確診罹患罕見疾病甘迺迪氏症。依循遺傳模式,小芳本身一定是帶因者,安排PGD胚胎著床前基因檢測,來迎接新生命的誕生。
遺傳模式
甘迺迪氏症為X染色體性聯隱性遺傳,性聯遺傳是指缺損的基因位於性染色體上,而隱性遺傳是必須一對(兩條)染色體皆有缺損才表現病徵。女性患者須有兩條缺陷的X染色體同時集中在一起,才會發病;若只有遺傳一條有缺陷的基因,則是無症狀的帶因者,女性帶原者所生育的子代中,50%的男孩會罹患這個疾病,而有50%的女孩會為帶原者。但在男性則只需一條缺陷基因即會產生症狀,男性患者所生育的子代中,所有的女兒則皆為帶原者。
主要好發於男性,且多在青壯年發病,女性則多為無症狀或症狀輕微的帶因者。疾病發生率約為1/50,000,大多在30~50歲。有些患者會有胸部發育、不孕和睪丸萎縮情形。病徵與漸凍人(amyotrophic lateral sclerosis)相似易被誤診,但兩者差異漸凍人通常進展快速,且會有上運動神經元退化的徵象,也不會有男性女乳症及睪丸萎縮的現象。則甘迺迪氏症進展是漸進且緩慢,發病後可活三、四十年,但通常發病二十年後可能會需要使用拐杖或輪椅。
病理
患者發病後的病程進展大多緩慢且症狀多變,早期常見症狀包括:雙手不自主顫抖、行走困難或快跌倒樣、肌肉痙攣及皮下肌束自發性震顫,而患者肌肉無力的狀況常從近端脊髓開始延伸,之後臉部、舌頭和喉頭的延髓肌肉,可能因無力而導致患者吞嚥與說話困難,也因此容易引發吸入性肺炎,最終可能得終生倚靠輪椅或四肢癱瘓。因為雄性激素蛋白質無法正常作用,使得男性患者常有雄性激素缺乏及雌激素過盛的情形,故常會有男性女乳症、精蟲數量低與不孕等生殖系統的問題。除此之外,也有部分患者會伴隨非胰島素依賴型糖尿病。
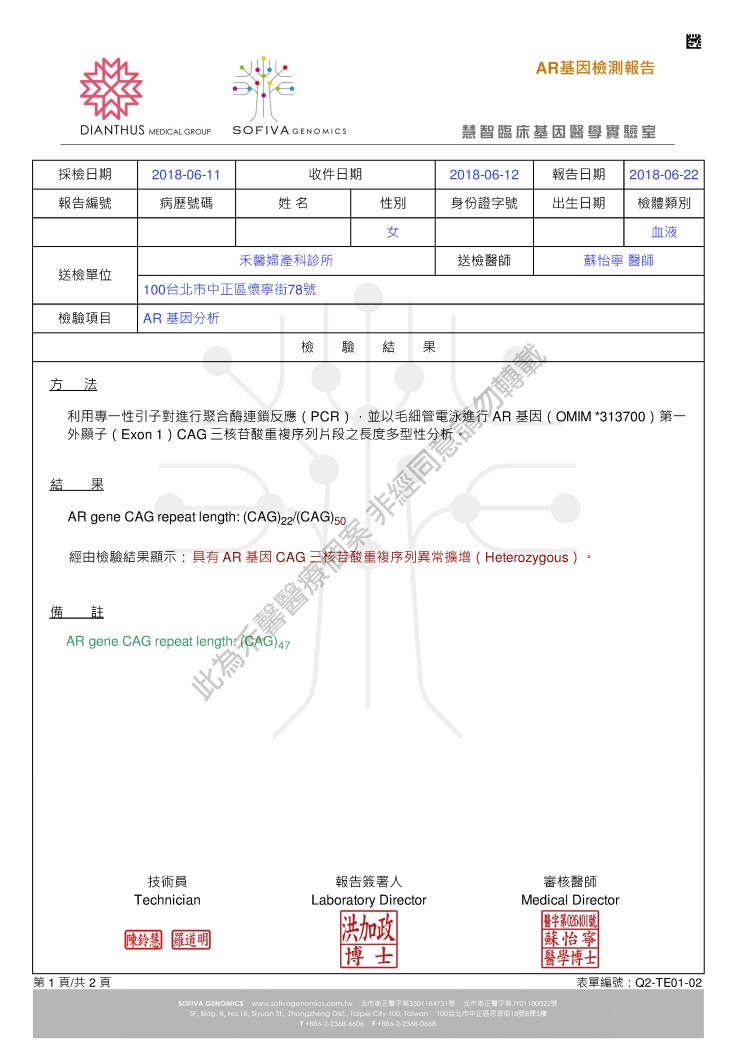
此為禾馨醫療個案 非經同意請勿轉載
個案圖譜



請問甘迺迪氏症帶因者如果已經懷孕,NIPS+有辦法檢測嗎?
讚讚
我也想問下這個
讚讚
我在105年經成大醫院確認是甘迺迪氏症患者,醫生說目前無藥可醫,我長期復健可是功效不彰手腳漸漸退化無力,冒昧請問博士還有其他治療方法嗎?國外有發展出新藥可治療嗎?謝謝
讚讚
我也是甘迺迪氏症患者目前有搜尋安基生技新藥AJ201,說是2022年下半年在美國做2期人體實驗,請問在國內有沒有推薦的醫學院可以做支持治療的
讚讚
只在美國測試
讚讚